
Laboratories
When the Division of Biologics Standards (DBS) was established in 1955, it centralized the biologics regulatory function at the National Institutes of Health (NIH) into one division, rather than having staff in multiple laboratories and institutes; however, for the first five years, the staff was still spread among multiple buildings on the NIH campus. In 1960, Building 29 was completed, and the staff was consolidated into one space. During the early 1960s, the laboratories and the sections within them were still being finalized and organized based on growing scientific knowledge and the demands of vaccine production and other biologics regulations. By 1968, the DBS had established the seven laboratories described below.
When the DBS was administratively transferred to the Food and Drug Administration (FDA) Bureau of Biologics in 1972, many of these labs remained the same, but under the title of Division instead of Laboratory. During the 1980s there was significant reorganization, and many laboratories changed names, combined with other laboratories, or were closed entirely.

The locations of the laboratories, if looking at the front elevation of Building 29 (shown above):
Laboratory of Bacterial Products
- 4th Floor (Rooms 420, 429, 431, 429A, 432, 418, 406, 416); left side of 4th floor
Laboratory of Biophysics and Biochemistry
- Rooms 305 and 306; right side of 3rd floor
Laboratory of Blood and Blood Products
- 1st floor (from 1960 to 1967) (Rooms 112, 107, 114, 111, 104, 100, 404, 400) and then the 2nd and 3rd floors (after 1967) (Rooms 318, 324, 326, 311, 225, 330, 219, 306); right side of 1st floor, then left side of 2nd and third floors
Laboratory of Control Activities
- 4th floor (from 1960 to 1967) (Rooms 400, 405, 416, 401, 402, 403); right side of 4th floor
Laboratory of Pathology
- Rooms 512 and 516; middle left side of 5th floor
Laboratory of Viral Immunology
- 2nd floor (from 1960 to 1967) (Rooms 212A, 229, 200, 208, 209, 230, 221, 311); most of 2nd floor
Laboratory of Virology and Rickettsiology
- 2nd and 3rd floors (from 1960 to 1967) (Rooms 318, 308, 332, 207, 216, 316, 330, 222, 214, 207); most of 3rd floor
Organizational Changes in CBER
In 1988, a new research division was established within CBER, the Division of Cytokine Biology. Cytokines are proteins that have multiple cellular functions, including effects on the immune system, antiviral activities, and the ability to differentiate between cells. Cytokine biology has the potential to further the development and review of therapies for diseases like AIDS and cancer.
In March 1989, CBER had six divisions (listed below), and each division had two to seven laboratories within it, totaling 29 laboratories related to biologics regulation at the FDA. Some laboratories had sections within them, much like DBS did in the 1960s. Many of these divisions are similar in their research and regulation roles to the 1960s DBS laboratories listed above.
Division of Product Quality Control
Research in this division focuses on improving existing quality control tests for biologics by finding ways to improve animal tests or to switch them to in vitro (test tube or cell culture) tests. The biological testing laboratory is within this division, and it has two sections: the reference reagents section and the animal testing and quality assurance section. The reference reagents section establishes the U.S. Reference, or standard preparation used by the FDA and manufacturers when performing official tests required for biologics. The animal testing and quality assurance section is responsibility for testing the potency and sterility of biological products and does safety tests on vaccines.
Division of Blood and Blood Products
The Division of Blood and Blood Products performs research on the preparation, preservation, and safety of blood and blood products, and methods for testing the safety, purity, potency and effectiveness of these products when used therapeutically. There are seven labs in this division: cell biology, transplantation biology, blood bank practices, retrovirology, hepatitis, plasma derivatives, and cell components.
Division of Virology
Vaccine testing and research is done by this division, including their four labs: DNA virus research, respiratory virus, pediatric diseases, and retrovirus.
Division of Cytokine Biology
This was the newest division of CBER in 1989, having only been formed in 1988. It was an outgrowth of the Division of Virology. Cytokines are proteins that have multiple cellular functions, including effects on the immune system, antiviral activities, and the ability to differentiate between cells. The division is responsible for conducting regulatory review and research on biological response modifier agents (medical treatments derived from substances naturally found in the body), which may hold promising treatments for AIDS, cancers, and infectious diseases. Laboratories under the Division of Cytokine Biology include cellular immunology, cell biology, and molecular immunology.
Division of Biochemistry and Biophysics
This division is an interdisciplinary group who studies molecular and cell biology, pharmacology and toxicology, biochemistry and biophysics, and combines basic science with regulatory activity. There are seven labs in this division: molecular immunology, biochemistry, analytical chemistry, biophysics, cellular and molecular biology, molecular pharmacology, and chemical biology.
Division of Bacterial Products
This division has seven labs within it, including pertussis, allergenic products, bacterial toxins, bacterial polysaccharides, mycobacteria and cellular immunology, mycoplasma, and cellular physiology.
Allergenic Products
Allergenic investigations by the FDA Bureau of Biologics in 1978 focused on standardized in vitro and in vivo assays of allergenic potency. The former tests under development included radial immunodiffusion, isoelectric focusing, and allergen coupling compared to radioactive or enzyme-labeled antibody. The in vivo assays under study were based on the skin tests used in allergic patients as well as the immunogenicity of allergenic extracts following their use in the immunotherapy of allergic disease.
Mycoplasma
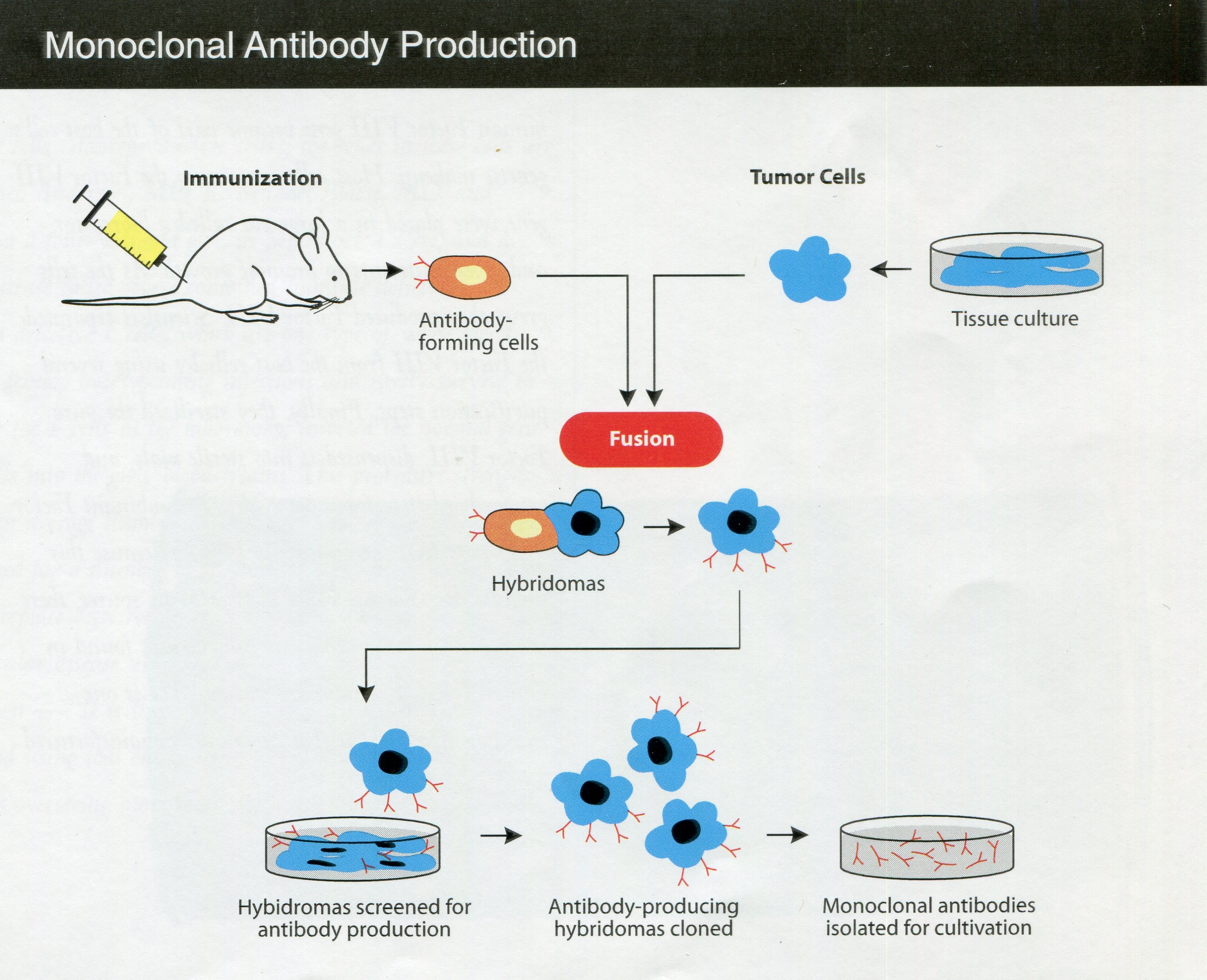
By the early 1980s, the FDA Bureau of Biologics had a laboratory on mycoplasma, but scientists had been investigating mycoplasmas since before the DBS moved into Building 29 in 1960. Mycoplasmas are single-celled bacteria lacking a cell wall and are able to penetrate other cells; they constituted a challenging source of contamination of cell cultures. A DBS regulation in 1960 required manufacturers to test for mycoplasmas in human viral products that relied on cell cultures. As mycoplasma tests advanced over the years, the testing rules changed accordingly. With the emergence of hybridoma technology to generate biological products (in which a target antigen is injected into a mouse, a resulting antibody-producing cell is harvested from the spleen, and the splenic cell fused with a tumor cell to produce the desired antibody), mycoplasmas found their way into the new products from this technology, such as monoclonal antibodies. The Bureau of Biologics’ rulemaking and research interests by the 1980s thus had shifted to controlling mycoplasma contamination of the products developed from this newer approach to biologics production.
When the biologics regulation function emerged after the many organizational changes at the FDA as CBER, it then included an Office of Establishment Licensing, an Office of Blood Research and Review, an Office of Vaccine Research and Review, an Office of Therapeutic Research and Reviews, and the Office of Compliance.
Office of Compliance
Compliance as it relates to Blood and Blood Products
Almost simultaneously with the transfer of biologics from the NIH Division of Biologics Standards to the FDA Bureau of Biologics in 1972 were two key changes in the regulation of blood products. First, all federally licensed blood banks had to test for the hepatitis associated antigen (HAA), a discovery within the previous decade, and exclude from donation any blood testing positive. Also, the FDA Bureau of Biologics licensed a new test for HAA, much more sensitive than previous tests, and Bureau staff continued their research into test improvements.
Inspections of intrastate blood collection establishments in 1974 indicated several widespread problems: slightly less than half the inspections revealed failure to adequately establish donor suitability, about the same percentage demonstrated problems with their blood collection techniques, and half exhibited faulty hepatitis testing. To get a better handle on the massive effort to regulate the blood and blood product industries, the Bureau of Biologics initiated a database, the Blood Establishment Inspection and Registration System—finalized in 1976—to monitor the thousands of annually renewed registrations and licenses, the biennial inspections of several thousand facilities, the compliance history and progress with the firms, and impact of regulatory changes.
In addition to these moves in the regulation of blood, the Bureau quickly proposed revisions to the licensing standards for blood plasma, the source for various products including gamma globulins, antihemophilic factor, and serum albumin. Also, as mentioned earlier, only about 500 interstate blood banks were regulated by the federal government; the 6000-7000 intrastate blood banks operated with little to no oversight. The Bureau invoked authorities under the 1938 Food, Drug, and Cosmetic Act to apply to all blood collection and processing centers. Blood banks and other blood collection facilities now had to be registered and inspected. Plasmapheresis centers (sites where blood is collected, the plasma is separated from the red blood cells, the latter are returned to the patient, and the plasma either treated and returned or repurposed) had to be licensed and inspected. Nearly 6000 establishments were registered in the first 15 months, and Bureau and Bureau-trained field office personnel inspected about 1000. The Bureau trained FDA field investigators such that the latter were able to take over the inspection responsibilities of the nearly 250 plasmapheresis facilities by 1977. Two years earlier, FDA for the first time revoked a biologics establishment license for cause, a recidivist plasmapheresis firm. In 1977, FDA pursued its first criminal prosecution of a plasmapheresis firm. In this case, the corporation was charged with providing false documents to FDA, intentional and non-intentional violations of the Food, Drug, and Cosmetic Act by adulterating and misbranding plasma, and violation of the Public Health Service Act by misbranding a biological product. A jury found the three charged officers of the firm guilty. Each was sentenced to 18-24 months in prison and their company fined $27,000. The plasmapheresis center and its ancillary facilities were enjoined and closed.

A significant change in oversight of blood came with the finalization of new good manufacturing practices for all blood banks, transfusion centers, and blood processing entities. Among other provisions, these required testing all donations for hepatitis, standards were established for tests given to patients prior to the administration of blood or a blood product, any fatality involving a blood donation or administration had to be reported to FDA immediately and required recordkeeping for blood products was extended. A retrospective analysis of plasma derivatives submitted since 1960 up to 1973 indicated that almost all lots of plasma protein fractions had the Hepatitis B Antigen.
In 1975, FDA issued a regulation requiring that all blood, plasma, or serum used for biological products must be tested by the latest, “third generation,” technology. This included reverse passive hemoagglutination and radioimmunoassay, which were 10 to 100 times more sensitive than the most common test required since 1972, counterelectrophoresis. Results from FDA’s contractual partner in evaluating the latest testing procedures, the New Jersey Department of Health, indicated that the new testing technologies would identify and thus eliminate from consideration as many as two times as many units of hepatitis-contaminated blood.

In mid-1986, Biologics suspended the licenses of four blood banks in Texas, Michigan, Missouri, and Kansas whose operations varied so significantly from required good manufacturing practices as to represent a danger to health. Each was inadequately supervised and the staff insufficiently trained, in addition to other problems. Lacking a license, they were not allowed to operate until FDA determined they were in compliance, and the Commissioner had the discretion to revoke their licenses in the absence of corrections.
Prompted by concerns with the transmission of the AIDS virus, FDA launched an intensive inspection program of all blood banks and plasma centers in 1988. Over 11 percent of the facilities were found to have significant enough violations to prompt a regulatory response. These problems included the processing of blood suspected of contamination with the AIDS virus, although there were no cases where blood or products confirmed to be so contaminated were released. During this program inspections of American Red Cross (ARC) blood banks in Nashville and Washington, D. C. indicated operational errors leading to the release of unsuitable units of blood and blood products, errors that continued despite corrections. So, in 1988 FDA and ARC signed a voluntary agreement in which the latter would improve operations across the nation by establishing closer oversight of regional operations by national headquarters, standardizing operating procedures, and monitoring more closely computerized information to prevent release of unsuitable blood.
Despite the 1988 voluntary agreement agency inspections of ARC facilities across the nation continued to present compliance failures, followed by formal regulatory notices and initiation of proceedings to revoke establishment licenses. In May 1993, ARC agreed to a consent decree, enforceable by court order, that stipulated a comprehensive series of changes to ensure the safety of the nation’s blood supply and the integrity of ARC’s blood program. Among the changes: establishing a clear line of managerial control over quality control in all regions; establishing a comprehensive quality control program; enhancement of a training program to be required—along with an annual competency review—for all staff engage in blood programs, improvement of its computer system, records management, and its policies for following up on reporting errors, accidents, and adverse events; and annual performance audits of each of its 47 blood program regions.

Other FDA Compliance and Enforcement
Recalls are an important part of regulating biological products. Some are initiated by the company; others proceed from FDA’s request. The Bureau of Biologics took part in its first biologics recall in FDA in 1973 when a Philadelphia firm issued a voluntary recall for immune serum globulin that was distributed in vials containing 25 percent less product than labeled. After the transfer to FDA, the Bureau utilized a new enforcement approach to violative biologics, employing FDA-supervised voluntary recalls. Their employment resulted in non-sterile serum albumin linked to multiple cases of septicemia, hypo-potent HAA, misbranded bacterial antigen, and other unlawful biological products being efficiently and thoroughly taken out of distribution and followed up by the agency.
Inspections and enforcement actions by the Bureau of Biologics grew such that by 1974 they conducted 800 inspections of establishments holding biologics licenses. Office of Biologics’ regulations called for at least one inspection annually for licensed establishments, though that was adjusted to a biennial basis in 1983 to give Biologics more flexibility to manage its resources and align better with inspection frequency for drug and medical device establishments. The Bureau was responsible for testing lots of antitoxins, vaccines, blood products, diagnostics, and other biologics prior to release. In its first full year in the FDA Bureau scientists tested over 6200 lots of these products and rejected 144. Often the problems were due to hypo-potency, but pyrogenicity, lack of sterility, and other issues were responsible as well. Nearly two dozen blood and allergenic products were recalled under Bureau supervision for pyrogenicity and other problems; one of these had led to a hepatitis outbreak. This outbreak, traced to up to three lots of plasma protein fraction in an Indianapolis hospital blood bank, was attributed to inadequate pasteurization, and all of the firm’s plasma protein fraction and normal serum h was recalled.
The agency issued over 300 Regulatory Letters to establishments in 1974—mostly to blood facilities. These official letters identify violations and indicate a deadline for correction before the party is subject to court proceedings. The Bureau of Biologics also obtained a decree for the destruction of improperly stored lots of influenza vaccine and tetanus toxoid and secured a permanent injunction against multiple facilities of a Florida plasmapheresis firm in major and repetitive violation of regulations for the collection and processing of plasma. Two years later FDA obtained the first permanent injunction against an intrastate blood bank, which enjoined it from operating in violation of the Federal Food, Drug, and Cosmetic Act. Inspections at an Ohio hospital blood bank on two occasions revealed serious deficiencies which threatened both blood donors and recipients, and each visit prompted increasingly severe warnings if non-compliance with regulations continued. After a third inspection revealed ongoing and hazardous operations outside of the regulations, the agency secured the permanent injunction, which allowed the hospital to operate only in emergency situations and to receive blood from outside sources.
Adverse event reporting has had a checkered history across all FDA medical product centers. In October 1994, CBER amended the biologics regulations to require that manufacturers of licensed biological products must report to FDA, within 15 working days, all serious and unexpected adverse events relating to their products; any significant increase in the frequency of serious but expected reactions; periodically, all other adverse experiences; and biologic distribution information.
Regulation of the biologics marketplace can take many different forms and must be equipped to pivot quickly to address countless threats to public health. For example, Congress appropriated $300,000 for FDA to hire 48 temporary investigators to help prevent the diversion of DBS-cleared lots of licensed Salk polio vaccine from legitimate distribution channels—those used by the six producers to ship vaccine to the states and then to public and private distributors. FDA provided the training, oversight, and on-site assistance for the temporary staff. Though unclear how long this program would last, the agency did not encounter any missing vaccine. A case from 2003 illustrated another turn the regulation of biologics could take. FDA’s Office of Criminal Investigations, established only shortly before, announced the discovery of counterfeit Procrit (epoiten alfa) in the marketplace. Some samples were contaminated with bacteria, and FDA laboratories found others completely lacking in active ingredient. Both the manufacturer and FDA alerted healthcare providers and patients as well to the problem and the lot numbers in question. Four months later criminal investigations announced three convictions in this case, though it was unclear if patients had been injured by the counterfeit product.


Office of Therapeutic Research and Reviews
The FDA Office of Therapeutic Research and Reviews included Divisions of Cytokine Biology, Cellular & Gene Therapies, Hematologic Products, Monoclonal Antibodies, Clinical Trial Design, and Application Review & Policy.
Organ and Tissue Transplantation
FDA issued the first license for human leukocyte typing sera in 1975, making it broadly available for use in identifying potential donors for organ transplants and transfusions. The following year the Bureau established a histocompatibility testing laboratory to provide quality assurance testing and to create reference standards for anti-human leukocyte typing serums proposed for commercial release. In 1982 the Bureaus of Biologics and the Bureau of Medical Devices agreed, and the FDA Commissioner approved that leukocyte typing sera would be delicensed and regulated as an in vitro diagnostic under the Medical Device Amendments of 1976.
Cytomegalovirus Immune Globulin Intravenous (CMV-IGIV) was licensed by FDA in 1990. Since an immune-suppressed organ recipient may be unable to fight off the effects of the virus, this action would help to protect kidney transplant patients from the effects of cytomegalovirus that may have been transmitted through the organ of CMV-positive donors.
In July 1986, FDA licensed the first therapeutic monoclonal antibody, Muromonab CD-3, to treat acute rejection of transplanted kidneys. The biologic bound to the T cell CD-3 antigen and rapidly dispatched the T lymphocytes.
In May 2005, new regulations took effect requiring human tissue firms to properly screen and test donors among other aspects of operations. These also provided for swift action to be taken by FDA in the interest of public health. Applying these new regulations early in 2006, FDA issued to a Ft. Lee, New Jersey, human tissue recovery firm an order to immediately cease all manufacturing operations. The agency monitored the recall of all their tissues to ensure completeness of the operation. So egregious were the deficiencies in the firm’s manufacturing practices, donor screening, record keeping, and other operations that, according to a senior agency official, “allowing the firm to manufacture would present a danger to public health by increasing the risk of communicable disease transmission.”
Therapeutic Research and Regulation
The FDA Bureau of Biologics scientists investigated a biological used to prevent tuberculosis, BCG vaccine, in animal models for possible application in cancer immunotherapy, following on from results from other researchers.
Late in 1980, Biologics scientists developed tests that might be applied to viral-based diseases of the central nervous system, including subacute sclerosing panencephalitis. They found a way to measure small amounts of viral antibodies in the cerebrospinal fluid (CSF). In a study published in Lancet involving a control group plus about two dozen suffering from schizophrenia, tests of CSF for eight common viruses, including measles, mumps, and herpes simplex, they found a substantial disproportion of cytomegalovirus in the CSF compared to serum—a suggestive link but certainly one needing further study.
The FDA Office of Biologics Research and Review (OBRR), what biologics was known as is in the 1980s, ramped up the facilities and understanding necessary to address the regulatory aspects of advances in genetic engineering, including nucleic acid sequencing and gene cloning technology. Guidelines soon followed for recombinant DNA products, such as monoclonal antibodies, which were finding their way into licensable commodities such as in vitro blood tests and in vivo diagnostics and therapeutics.
FDA licensed two recombinant interferons, Interferon alfa-2a and Interferon alfa-2b for the treatment of hairy cell leukemia in 1986, the first of their kind. They were shown to arrest or regress the disease, reduce disease complications, and lengthen survival, with relatively untoward side effects. OBRR continued its research into the structure and function of interferons, and more than a dozen were awaiting further study, though their therapeutic promise had still not been assessed. Discovered decades earlier as naturally occurring anti-infective proteins in the body, recombinant DNA techniques made sufficient quantities available for research and therapeutic application.
The Center for Biologics Evaluation and Research (CBER) licensed recombinant Human Tissue Plasminogen Activator on November 13, 1987, to limit heart damage by dissolving blood clots if administered soon after a myocardial infarction. Two years later the Center approved Eminase (anistreplase), another bioengineered blood clot dissolving product to help prevent damage following a heart attack. It was designed to afford quicker administration and circulate in the system longer than other treatments in this class.
On June 1, 1989, CBER approved epoetin, a treatment for anemia due to chronic kidney failure. This was a copy of erythropoietin, the protein responsible for stimulating reproduction and growth of red blood cells in bone marrow. Blood transfusions were the typical approach to treating severe anemia, though those came with a host of risks and problems. Though this approval was limited in scope, other studies were underway to assess epoetin in several other applications linked to anemia, such as anemia-induced cancer chemotherapy and AIDS.
Botulinum Toxin A, an injectable treatment for eye conditions such as strabismus, was licensed by CBER in 1989.
In 1990, CBER approved the use of live bacteria to treat bladder cancer. BCG Live (after bacillus Calmette-Guérin), a freeze-dried suspension, was delivered directly to the bladder via a catheter to treat pathogenic cells lining the inner surface of the organ—and thus a treatment designed for earlier stages of the illness. The patient retained the product for two hours before voiding, and the treatment would be repeated for the next several weeks or months.

CBER licensed the first drug genetically engineered product to treat adult kidney cancer specifically, aldesleukin, in May 1992. The drug produced some serious side effects, to an extent that it had to be administered intravenously only in a hospital by clinicians experienced in cancer chemotherapy. However, at this time about 10,000 patients died of renal cancer annually, and aldesleukin reduced tumors in 15 percent of those treated, and in 4 percent tumors disappeared for nearly two years.
Toward the end of 1992 FDA licensed satumomab pendetide, a diagnostic imaging agent for ovarian or colorectal cancer patients that detected proteins found in high concentrations on the surface of these types of cancer. The agent was prepared by altering a monoclonal antibody to bind with a radioactive element, the two combined just prior to administering the preparation to the patient. However, it was not a screening test but could be used in tandem with CT scanning.
In July 1993, CBER issued the first license for a biologic treatment for multiple sclerosis, Interferon beta-1b. This also was the first biotechnological product licensed under FDA’s accelerated approval regulations. Since there was no approved therapy for MS this was designated a priority review. At the time there were an estimated 250,000-350,000 cases in the U. S. Also, though a biologic was subject to the provisions for licensing under the law, the review was carried out collaboratively by CBER and the Center for Drug Evaluation and Research, which provided clinical review expertise. A two-year study of 338 patients showed that interferon, in comparison to a placebo, decreased flare-ups by 30 percent and reduced brain lesions as captured by MRI scans by 20 percent. However, it did not change the course of the disease. The Peripheral and Central Nervous System Advisory Committee recommended approval of Interferon beta 1-b for relapsing-remitting MS by a 7-2 vote. Four months after the advisory committee vote CBER licensed the biologic. Under accelerated approval regulations the firm had to carry out post-marketing studies to investigate interferon’s slowing or preventing progress of the disease.
In December 1997, CBER scientists Drs. Tamas Oravecz and Michael Norcross and several other researchers in the Division of Hematologic Products, along with Dr. Mark Gorrell of the University of Sydney, published results of research they conducted that uncovered how immune cells are directed to respond to infections. A protein known as CD26, found on the surface of certain white blood cells, could activate or inactivate chemokines. The authors looked at the relationship between CD26 and several chemokines and found that “target cell recruitment into inflammatory sites may depend both on the extent of CD26 activity on chemokines and on the maturational status of the responding cells.” This new understanding of CD26, the authors believed, suggested a pathway toward new therapies for various infections and other diseases where chemokines have a role.
CBER licensed Herceptin (trastuzumab) in September 1998, a monoclonal antibody that represented a new approach to treatment of metastatic breast cancer. Bioengineered from an altered antibody in a mouse, the monoclonal antibody bound to HER2, a protein that regulates cell growth that is found on certain normal cells. This ability to bind to the protein enabled the antibody to interfere with tumor cell growth. In metastatic breast cancer, about 30 percent of the tumors express excess HER2, so this would be useful only in patients with tumors of that character. FDA approved Herceptin alone for those who received little benefit from chemotherapy or as a first-line treatment when used in combination with paclitaxel.
CBER and the National Cancer Institute (NCI) began a collaborative research and clinical project in 2001, the Clinical Proteomics Program, to make a comprehensive study of all proteins in living cells and apply this to the clinical care of cancer patients. Drs. Emanuel Petricoin in CBER and Lance Liotta of NCI led the effort, which was funded for three years about $1 million annually. The two groups had already developed new or improved analytical technology for the purpose, have identified more than 130 different proteins found in several types of cancer. Using the latest microscopic procedures to biopsy cells pre- and post-treatment would allow the team to analyze the impact of different therapies on tumor protein patterns. Among other benefits the Program hoped to achieve was earlier ability to diagnose cancer, a better sense of toxic and beneficial effects from the lab before clinical application, and improved understanding of tumors at the protein level, and better treatments for cancer patients. The following year the Proteomics collaborative reported preliminary results in The Lancet on diagnostic developments they uncovered by using protein patterns found in normal serum and that collected from an ovarian cancer patient.


Dr. Lance A. Liotta, at left, and Dr. Emanuel F. Petricoin, at right. FDA History Office
The 2007 Food and Drug Administration Amendments Act gave the agency authority to require a manufacturer to submit and carry out a Risk Evaluation Mitigation Strategy (REMS) for selected biologics or drugs to ensure a product’s benefits outweigh its risks as a condition of the product’s approval. FDA issued its first guidance under this authority in 2009. The elements of a REMS included a variety of communication devices to help inform the patient about using the therapy, such as medication guides and patient package inserts; a communication plan for the healthcare provider; and other elements to ensure safe use of the product aimed at the prescriber, dispenser, and the patient. The draft guidance addressed the format and content of the REMS, its assessment, and a timetable for submitting it.
Overview
Content Tools
ThemeBuilder