
Exhibit Overview
Biologics Regulation and Research: The People and Work of Buildings 29 & 29A
"Science and Peace will triumph over ignorance and war, nations will unite, not to destroy, but to build, and the future will belong to those who will have done the most for suffering humanity"
- —Louis Pasteur
Buildings 29 and 29A are nationally significant to the history of medicine and public health because within the laboratories of Buildings 29 and 29A, the National Institutes of Health (NIH) and then the Food and Drug Administration (FDA) staff helped to conquer some of the deadliest infectious diseases. In their regulatory role, they had the national responsibility to license vaccines, antitoxins, blood products, and other biologics to ensure their safety and effectiveness. To support this mission, they did scientific research which resulted in the development of important standards and even new vaccines. Some of the most well-known scientists of the 20th century worked in these buildings, as well as the key administrators and others who supported their work and the mission of biologics regulation and 22 of them are profiled here in the biographies section. This online exhibition will share information about the research, regulations, and work conducted in these two laboratory buildings between 1960 and 2014. A variety of diseases, vaccines, and other biologics are discussed, with links to articles and other websites for further research. While the buildings are no longer in use, the legacy of their exemplary staff and their important work endures.

What are Biologics?
Biologics are biological products made to prevent or treat a disease. They include vaccines, blood and blood products, allergenics, somatic cells, gene therapy, tissues, and recombinant therapeutic proteins.
The Biologics Control Act (1902)
The Biologics Control Act or the Virus Toxin Act, was passed in 1902 after 13 children died in St. Louis from tetanus, which had contaminated the diphtheria antitoxin they had received in 1901. Dr. Joseph J. Kinyoun of Marine Hospital Service’s Hygienic Laboratory (the ancestor of the Division of Biologics Standards [DBS] and the NIH more broadly) recognized the danger posed by the availability of unstandardized and poorly tested diphtheria antitoxins. Kinyoun wanted to establish an independent testing laboratory, however it took an incident in 1901 in St. Louis to get Congress to act, when 13 children who had received the antitoxin died of tetanus. An investigation discovered that one of the horses used in the manufacture of the antitoxin had live tetanus organisms in its blood and was the source of the infection.
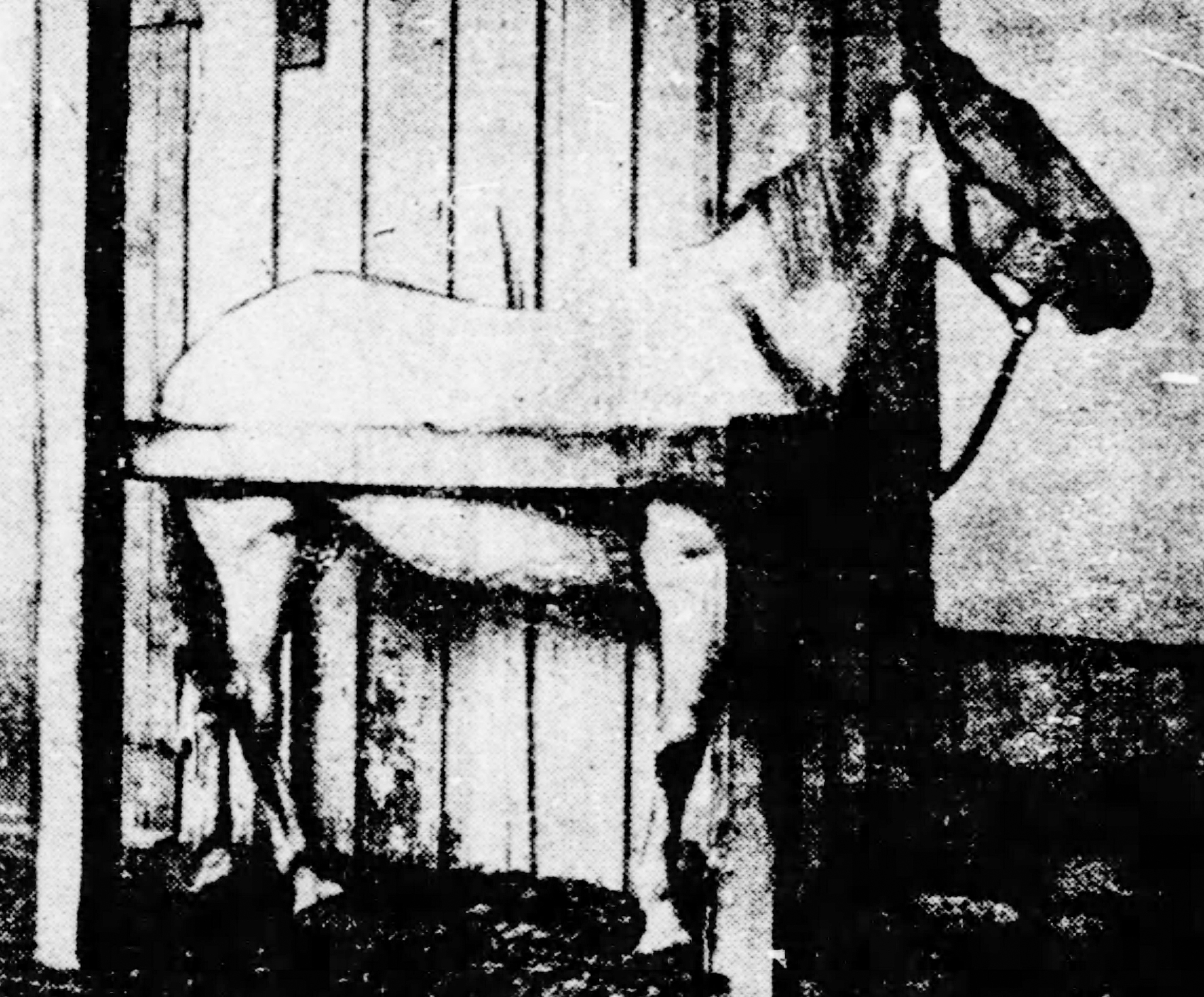
Congress then introduced two bills, both enacted on July 1, 1902. One bill established the Public Health and Marine Hospital Service, the other authorized this new service to regulate the sale and transportation of any virus, therapeutic serum, toxin, or analogous product in interstate commerce or from a foreign country. All establishments engaged in the sale, barter, or exchange, or offering for sale, barter, or exchange of biological products in interstate commerce must be licensed, and each product must be licensed individually. Each licensee must be inspected annually by agents and officers of the Federal government. The second act is known as the Biologics Control Act or the Virus-Toxin Act. This act has been amended by Congress several times to incorporate additional product classes, including blood, blood components, and blood derivatives. Today the regulated biologics include vaccines, blood and blood products, allergenics, somatic cells, gene therapy, tissues, and recombinant therapeutic proteins.

The Federal Food and Drugs Act (1906)
In 1906, the Federal Food and Drugs Act made illegal adulterated and misbranded foods and drugs, although it did not specifically mention biological drugs. Unlike the 1902 Biologics Control Act, which passed easily in Congress, the 1906 Act sparked serious debate for decades. The 1906 Act had some shortcomings, which led to the passage of the Sherley Amendment in 1912 which prohibited labeling medicines with false therapeutic claims intended to defraud the purchaser.
The Federal Food, Drug, and Cosmetic Act (1938)
To strengthen consumer protection, the 1906 Act was replaced with the Federal Food, Drug, and Cosmetic Act of 1938. 107 people had died after consuming “Elixir Sulfanilamide” which was a misbranded commercial product that was made with toxic diethylene glycol instead of alcohol. Under the 1938 Act, a biological product was considered a drug. The 1938 Act did not modify or supersede the 1902 Biologics Control Act. After 1938, the appropriate provisions of the both the 1902 and 1938 acts were used to regulate biologics.
In 1951, the Durham-Humphrey Amendment to the Federal Food, Drug, and Cosmetic Act of 1938 was passed and required that the type of drugs that could not be used safely without medical supervision had to be sold “by prescription only.”
In 1962, the Kefauver-Harris Amendments to the Federal Food, Drug, and Cosmetic Act of 1938 were passed after thalidomide, a sleeping pill used widely in Europe, was found to cause birth defects. Thalidomide had not been marketed in the US thanks to the FDA, but it had been used in a pharmaceutical company’s investigational trial and 624 of the 20,000 American participants were pregnant women. There were 17 documented cases of birth defects caused by thalidomide in the US. The 1962 amendments strengthened the regulations for drug safety and for testing drugs in clinical trials. They also required manufacturers to provide “substantial evidence” that their drugs were effective for their intended use and that the drugs had to be produced using “good manufacturing practices,” which required inspections of manufacturers every two years and yearly registration. The 1962 amendments also applied to blood banks.
The Public Health Service Act (1944)
In 1944, laws related to the Public Health Service (PHS) were revised and consolidated into one act, which helped to define medical research in the post-World War II era. The 1902 Biologics Control Act was incorporated into the 1944 Act. One change was that the Laboratory of Biologics Control was authorized to license biological products as well as the establishments where they were produced. The 1944 Act also provided them authority to manufacture biologics if the need arose.
Division of Biologics Standards
In 1955, the Division of Biologics Standards (DBS) was formed, as an independent entity at the NIH, but as the continuation of a biologics regulatory function that had existed in what is now known as the NIH since 1902. The predecessor of the DBS was known as the Division of Biologics Control, formed in 1937, whose name was changed to the Laboratory of Biologics Control in 1944, and its staff were spread out amongst multiple buildings on the NIH campus. The DBS was responsible for establishing and maintaining standards of quality and safety of all biological products. The safety, purity, and potency of all biologics must be established before the product was licensed by DBS.
The DBS was formed in the wake of the “Cutter Incident.” In 1955, the year following a field trial that showed the Salk inactivated (killed) polio vaccine to be safe and effective, the DBS licensed several firms to produce the vaccine. One firm, Cutter Laboratories, accidentally released vaccine that retained live polio virus, resulting in 260 paralytic cases of the disease, a disaster that caused panic among parents and scientists alike. The vaccine from Cutter Laboratories was pulled from the market on April 27, 1955, but the damage had already been done. The “Cutter Incident” became a defining moment in the history of vaccine manufacture and government oversight of vaccines. It occurred because the rigorous safety precautions used in the field trials were not required, at the time, for the production of commercially-produced licensed vaccines. The government realized it needed to strengthen its role in biologics regulation. The “Cutter Incident” led directly to the transfer of the regulation of biologics from the Laboratory of Biologic Controls to the DBS, a new independent entity with NIH.
In 1960, Building 29 opened, providing a consolidated space where the staff of DBS could work together. The 1950s through the 1970s were exciting and busy times for biologics regulation, as vaccine research flourished, new scientific advances in tissue culture were made, and important changes were made for regulating blood and blood products. This necessitated the construction of Building 29A in 1967, to provide an annex of laboratory space for the DBS staff.
Transfer to FDA and Biological Products Review
The legislative history leading to the codification of the 1902 Biologics Control Act into the 1944 Public Health Service Act included a proposal that biological products have ‘efficaciousness’ in addition to the safety, potency, and purity requirements already in the statute. However, the proposed additional requirement of efficaciousness was not carried into the 1944 Act. In March 1972 the Government Accounting (later called Accountability) Office (GAO) reported that DBS had not required effectiveness of biological products despite the fact that they were also ‘drugs’ under the statutory definition, and drugs had to be effective as well as safe under the 1962 Amendments to the Food, Drug, and Cosmetic Act. In the same month NIH announced its intention to review all of its biological products and issued a call for substantial evidence of effectiveness, the standard that was applied to drugs. During a May 1972 Congressional hearing Department of Health, Education, and Welfare (DHEW) Secretary Elliott Richardson announced that DBS should be transferred to FDA, and the following month he did just that.
In February 1973 FDA finalized plans to conduct a systematic efficacy review of all licensed biological products, modeled on its review of over-the-counter drug products that had been launched the previous year. Commissioner Charles Edwards appointed six advisory panels of experts that would review the evidence and assign each biological product to one of the following categories: safe and effective; unsafe and/or ineffective; requiring further study but to remain available in the interim; and lacking sufficient data to be determined safe and effective and recommended for market removal. Much as the case with new drugs, the proof of effectiveness would be expected to derive from adequate and well-controlled investigations. And much like the case with drugs, the review lasted decades. Along the way, though, some licenses have been revoked, such as in 1977 when FDA announced its intention to revoke the licenses of eight skin tests for tuberculosis, diphtheria, and other diseases and conditions.
Center for Biologics Evaluation and Research
The FDA Center for Biologics Evaluation and Research (CBER) is the home of biologics regulation today. When DBS was administratively transferred from the NIH to the FDA, it was called the Bureau of Biologics. During the 1980s and 1990s there was significant organizational change in biologics at the FDA. In 1982, the Bureau of Biologics merged with the Bureau of Drugs to form the National Center for Drugs and Biologics (NCDB). In 1983, the biologics component was renamed the Office of Biologics Research and Review (OBRR) within the Center for Drugs and Biologics (CDB). In 1988 more changes happened when the CDB separated into two centers: the Center for Biologics Evaluation and Research (CBER), which was formerly the OBRR, and the Center for Drug Evaluation and Research (CDER). CBER has remained the name for the biologics regulation team at the FDA ever since. In 2002, some functions of CBER were transferred to CDER, to allow CBER to focus on regulating vaccines, gene therapy, tissue transplantation, and blood safety. FDA’s review of new pharmaceutical products, which had been partially done by both CBER and CDER in the past, would be consolidated to CDER.
Architectural Documentation
Historic American Buildings Survey (HABS) architectural documentation of Buildings 29 and 29A at the NIH is part of the efforts to mitigate the adverse effects of the planned demolition of the buildings. The survey and fieldwork were completed July 12–14, 2021. The HABS materials will be available on the Library of Congress website in addition to portions of them being presented in this exhibition. This online exhibition on Buildings 29 and 29A and the research conducted there is also a part of the mitigation process.
The FDA Center for Biologics Evaluation and Research (CBER) moved from the NIH to the FDA White Oak Campus in 2014. This move completely vacated Buildings 29, 29A, and the circa-1994 Building 29B. Building 29B has been reoccupied by NIH staff, following moderate renovation. NIH completed feasibility studies and determined in 2020 that it is not viable to reuse Buildings 29 and 29A, and demolition is planned. As these are historic buildings on Federal property, Section 106 of the National Historic Preservation Act (NHPA) and its implementing regulations (36 CFR §800) must be followed. As part of the Section 106 process, consultation with various parties, including local government, historical societies, and the public place before a Memorandum of Agreement (MOA) was entered into between the NIH and the State Historic Preservation Office, the Maryland Historical Trust (MHT), since demolition of buildings is an adverse effect to historic properties. This online exhibition and the aforementioned HABS architectural documentation are the result of that agreement. While Buildings 29 and 29A are no longer in use, the legacy of their exemplary staff and their important work endures.
Overview
Content Tools
ThemeBuilder