
Introduction
Pain is a universally known and feared human condition, yet surely it is one of the least understood. Within this century it has been a philosophic problem, an enigma of neurophysiology, a psychological puzzle, a challenge to anesthesiologists, oncologists, nurses, clinicians of all kinds; and much of what we now think we know about pain still seems contradictory and paradoxical.
Pain and the Dentist

Here is another puzzle: how did the most versatile and productive pain research unit at NIH establish itself in the National Institute of Dental Research?
The story begins in the late 1950s in the dentist's office. The mass screenings of the World Wars had revealed the failure of many Americans to seek regular dental care and the disastrous consequences that followed. The American Dental Association, the National Institute of Dental Research (NIDR; renamed the National Institute of Dental and Craniofacial Research (NIDCR) in 1999), and other authorities actively promoted regular dental visits, especially for children. But increased numbers of visits only pointed to the underlying problem: many Americans associated dentistry with pain and faced each appointment with dread.
Seeking to alleviate patient fears, dentists offered general anesthesia for many routine procedures.
It is estimated," NIDR researcher Edward Driscoll wrote in 1960, "that in many parts of the country...there are nearly as many general anesthetics administered in dental offices as there are in the local hospitals."
- –NIDR Annual Report, 1960
 Edward Driscoll
Edward Driscoll
But this well-meant practice drew fire from anesthesiologists and health authorities, on several grounds. Dentists, even oral surgeons, rarely had formal training in anesthesiology or experience with many of the new agents introduced in the 1950s and 1960s. Even if they had, it was often impossible for one person both to perform the operative procedure and to monitor the patient's physiological status. The difficulty of monitoring was further complicated by the lack of information about the side effects of such new agents as sodium pentothal and sodium methohexital -- a particularly dangerous ignorance in light of the fact that patients generally left the dentist's office within an hour after the procedure. For all these reasons, dental anesthesia was condemned as unsafe, although there were few documented cases of patient injury.
Problems with general anesthesia in dentistry, 1960
Lack of training and experience
Lack of staff
Lack of data about agents used

Cycle of Fear and Pain. From Raymond A. Dionne, Pain Control in Dentistry: The Basis for Rational Therapy. Compendium of Continuing Education in Dentistry v. 6 (1985): 16.
Edward Driscoll of NIDR's1 Oral Medicine and Surgery Section began conducting studies of dental anesthesia in 1957. His aims were: to establish the necessary baseline physiological data; to evaluate the effects of stress on the dental patient; and to find the best methods of alleviation. With his associates, he performed full mouth extractions on more than 1200 patients, and collected readings for pulse, blood pressure, respiration, arterial oxygen levels, EEG, and EKG. Edward Driscoll. Photograph courtesy of NIDCR Public Information Office.
The results of Driscoll's studies in the late 1950s and early 1960s provided the first data on the efficacy and risks of dental anesthesia; unfortunately, much of the data were never written up for publication and the records were finally lost to the need for space when the Pain Branch expanded in the 1970s.
Ron Dubner and the "Lone Rangers"
In 1964, Ronald Dubner returned to NIH from the University of Michigan. Dubner had worked in the Clinical Center from 1958 to 1961 as a young Dental Officer in the Commissioned Corps; there he had discovered his interest and talent for research, and successfully applied for NIH support for his Michigan doctoral studies in neurophysiology. Seymour Kreshover , Scientific Director of the Institute, was a major supporter of the young researcher and his plans to establish a basic neuroscience laboratory at NIDR.
Dubner set up his lab in the basement of Building 30 and hired Fred Brown, a young electrical engineer, to put together the physiological recording apparatus he needed. He began recording from nerve cells in the brain and the trigeminal nucleus that are excited by visual and auditory stimuli.
How did he relate these studies to the mission of the Dental Institute? Through the problem of dental pain. Dubner argued that an understanding of the mechanisms of pain, and the development of methods for its management and control, had to be based on a thorough knowledge of how sensory information was processed. His early work showed that the first central relay nucleus in the trigeminal system was the site of significant modifications of sensory information; and that the association cortex in the brain was possibly the site where multiple sensory inputs converged.
The trigeminal system is the medullary section of the spinal dorsal horn which processes sensory information from the face and cranium.
Dubner's was among the very first research projects to accept the challenge laid down by Melzack and Wall's gate control model (1965) to describe the complex sensory, cognitive, and affective components of clinical pain as physically embodied in an interactive nervous system.
There were several other basic scientists working in Building 30 at the time: Bruce Dow and Barry Sessle in Dubner's lab; Steve Gobel a neuroanatomist who was interested in electron microscopy studies of the trigeminal system; and a group of microbiologists. When Kreshover became Director of NIDR in 1966, Richard Gruelich , the new Scientific Director, decided to build a basic science unit around these "lone rangers", which was named the Physiology Section. The following year, Gobel, Dubner, and his team were separated from the microbiologists and designated the Neural Mechanisms Section.
Dubner and his collaborators had demonstrated by 1969 that both ascending nerve fibers from the skin and descending fibers from the brain were synaptically connected to the dorsal horn of the spinal cord (the nucleus caudalis, or the "nerve center" of the trigeminal system). This connection suggested the presence of "a rapidly conducting feedback loop", which could act to enhance or alleviate information about sensory stimuli.
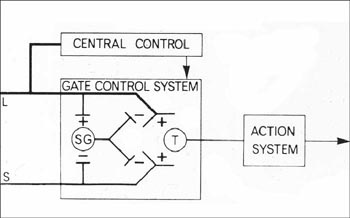
The Gate Control Model - Reprinted by permission - Ronald Melzack and Patrick Wall, Pain mechanisms: a new theory. Science v. 150 (1965): 975. Copyright AAAS, http://www.sciencemag.org

Seymour Kreshover

Patrick Wall [l] and Ronald Melzack [r]

Ron Dubner [l] and Barry Sessle [r] with colleague Eigild Moller at International Association for Dental Research, mid-1970s
References
- The National Institute of Dental Research (NIDR) was renamed the National Institute of Dental and Craniofacial Research (NIDCR) on October 21, 1998.
Overview
Content Tools
ThemeBuilder