
Introduction
How can you tell that a patient has a previously unknown disease?
How do you care for someone with a mysterious illness–and protect yourself as well?
How do you investigate the cause of a new disease, and find ways to treat and prevent it?
In Their Own Words documents how NIH researchers answered such questions when asked to recall the early days of HIV/AIDS. In launching this web site, we commemorate the 20-year struggle to confront the deadly HIV/AIDS pandemic.
Cold Spring Harbor Laboratory hosted the symposium: HIV/AIDS Research: Its History & Future, October 13-16, 2016. To see videos of the speakers, visit: History of HIV/AIDS Research.
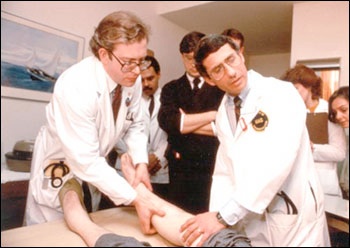
Drs. Lee Hall (left) and Anthony S. Fauci (right) examine participant in an early AIDS study

A crystal of the anti-AIDS drug zidovudine (AZT), viewed under polarized light

Drs. Thomas Folks and Guido Poli discuss their AIDS research

The NAMES Project AIDS quilt, representing people who have died of AIDS, in front of the Washington Monument
Overview
Content Tools
ThemeBuilder